Processing Whole Genome Sequences
1. A Brief Introduction to Arvados
Arvados is an open source platform for managing, processing, and sharing genomic and other large scientific and biomedical data. Arvados helps bioinformaticians run and scale compute-intensive workflows. By running their workflows in Arvados, they can scale their calculations dynamically in the cloud, track methods and datasets, and easily re-run workflow steps or whole workflows when necessary. This tutorial walkthrough shows examples of running a “real-world” workflow and how to navigate and use the Arvados working environment.
When you log into your account on the Arvados playground (https://playground.arvados.org), you see the Arvados Workbench which is the web application that allows users to interactively access Arvados functionality. For this tutorial, we will largely focus on using the Arvados Workbench since that is an easy way to get started using Arvados. You can also access Arvados via your command line and/or using the available REST API and SDKs. If you are interested, this tutorial walkthrough will have an optional component that will cover using the command line.
By using the Arvados Workbench or using the command line, you can submit your workflows to run on your Arvados cluster. An Arvados cluster can be hosted in the cloud as well as on premise and on hybrid clusters. The Arvados playground cluster is currently hosted in the cloud.
You can also use the workbench or command line to access data in the Arvados storage system called Keep which is designed for managing and storing large collections of files on your Arvados cluster. The running of workflows is managed by Crunch. Crunch is designed to maintain data provenance and workflow reproducibility. Crunch automatically tracks data inputs and outputs through Keep and executes workflow processes in Docker containers. In a cloud environment, Crunch optimizes costs by scaling compute on demand.
Ways to Learn More About Arvados
- To learn more in general about Arvados, please visit the Arvados website here: https://arvados.org/
- For a deeper dive into Arvados, the Arvados documentation can be found here: https://doc.arvados.org/
- For help on Arvados, visit the Gitter channel here: https://gitter.im/arvados/community
2. A Brief Introduction to the Whole Genome Sequencing (WGS) Processing Tutorial
The workflow used in this tutorial walkthrough serves as a “real-world” workflow example that takes in WGS data (paired FASTQs) and returns GVCFs and accompanying variant reports. In this walkthrough, we will be processing approximately 10 public genomes made available by the Personal Genome Project. This set of data is from the PGP-UK (https://www.personalgenomes.org.uk/).
The overall steps in the workflow include:
- Check of FASTQ quality using FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/)
- Local alignment using BWA-MEM (http://bio-bwa.sourceforge.net/bwa.shtml)
- Variant calling in parallel using GATK Haplotype Caller (https://gatk.broadinstitute.org/hc/en-us)
- Generation of an HTML report comparing variants against ClinVar archive (https://www.ncbi.nlm.nih.gov/clinvar/)
The workflow is written in Common Workflow Language (CWL), the primary way to develop and run workflows for Arvados.
Below are diagrams of the main workflow which runs the processing across multiple sets of fastq and the main subworkflow (run multiple times in parallel by the main workflow) which processes a single set of FASTQs. This main subworkflow also calls other additional subworkflows including subworkflows that perform variant calling using GATK in parallel by regions and generate the ClinVar HTML variant report. These CWL diagrams (generated using CWL viewer) will give you a basic idea of the flow, input/outputs and workflow steps involved in the tutorial example. However, if you aren’t used to looking at CWL workflow diagrams and/or aren’t particularly interested in this level of detail, do not worry. You will not need to know these particulars to run the workflow.


Ways to Learn More About CWL
- The CWL website has lots of good content including the CWL User Guide: https://www.commonwl.org/
- Commonly Asked Questions and Answers can be found in the Discourse Group, here: https://cwl.discourse.group/
- For help on CWL, visit the Gitter channel here: https://gitter.im/common-workflow-language/common-workflow-language
- Repository of CWL CommandLineTool descriptions for commons tools in bioinformatics:
https://github.com/common-workflow-library/bio-cwl-tools/
3. Setting Up to Run the WGS Processing Workflow
Let’s get a little familiar with the Arvados Workbench while also setting up to run the WGS processing tutorial workflow. Logging into the workbench will present you with the front page. This gives a summary of your projects in your Arvados instance (i.e. the Arvados Playground) as well as a left hand side navigation bar, top search bar, and help, profile settings, and notifications on the top right. The front page will only give you information about projects and activities that you have permissions to view and/or access. Other users’ private or restricted projects and activities will not be visible by design.
3a. Setting up a New Project
Projects in Arvados help you organize and track your work – and can contain data, workflow code, details about workflow runs, and results. Let’s begin by setting up a new project for the work you will be doing in this walkthrough.
To create a new project, go to the Projects dropdown menu and select the “+NEW” button, then select “New project”.

Let’s name your project “WGS Processing Tutorial”. You can also add a description of your project by typing in the Description – optional field. The universally unique identifier (UUID) of the project can be found in the URL, or by clicking the info button on the upper right-hand corner.


If you choose to use another name for your project, just keep in mind when the project name is referenced in the walkthrough later on.
3b. Working with Collections
Collections in Arvados help organize and manage your data. You can upload your existing data into a collection or reuse data from one or more existing collections. Collections allow us to reorganize our files without duplicating or physically moving the data, making them very efficient to use even when working with terabytes of data. Each collection has a universally unique identifier (collection UUID). This is a constant for this collection, even if we add or remove files — or rename the collection. You use this if we want to to identify the most recent version of our collection to use in our workflows.
Arvados uses a content-addressable filesystem (i.e. Keep) where the addresses of files are derived from their contents. A major benefit of this is that Arvados can then verify that when a dataset is retrieved it is the dataset you requested and can track the exact datasets that were used for each of our previous calculations. This is what allows you to be certain that we are always working with the data that you think you are using. You use the portable data hash of a collection when you want to guarantee that you use the same version as input to your workflow.

Let’s start working with collections by copying the existing collection that stores the FASTQ data being processed into our new “WGS Processing Tutorial” project.
First, you must find the collection you are interested in copying over to your project. There are several ways to search for a collection: by collection name, by UUID or by portable data hash. In this case, let’s search for our collection by name.
In this case it is called “PGP UK FASTQs” and by searching for it in the “Search” box. It will come up in the search results and you can navigate to it by clicking on the name. You would do similarly if you would want to search by UUID or portable data hash.
Now that you have found the collection of FASTQs you want to copy to your project, you can simply click the “Make a copy” button in the toolbar and select your new project to copy the collection there. You can rename your collection whatever you wish, or use the default name on copy and add whatever description you would like.
We want to do the same thing for the other inputs to our WGS workflow. Similar to the “PGP UK FASTQs (ten genomes)” collection there is a collection of inputs entitled “WGS Processing reference data” and that collection can be copied over in a similar fashion.
Now that we are a bit more familiar with the Arvados Workbench, projects and collections. Let’s move onto running a workflow.
4. Running the WGS Processing Workflow
In this section, we will be discussing three ways to run the tutorial workflow using Arvados. We will start using the easiest way and then progress to the more involved ways to run a workflow via the command line which will allow you more control over your inputs, workflow parameters and setup. Feel free to end your walkthrough after the first way or to pick and choose the ways that appeal the most to you, fit your experience and/or preferred way of working.
4a. Interactively Running a Workflow Using Workbench
Workflows can be registered in Arvados. Registration allows you to share a workflow with other Arvados users, and let’s them run the workflow by clicking the “+NEW” button and selecting “Run a workflow” on the Workbench Dashboard or on the command line by specifying the workflow UUID. Default values can be specified for workflow inputs.
We have already previously registered the WGS workflow and set default input values for this set of the walkthrough.
Let’s find the registered WGS Processing Workflow and run it interactively in our newly created project.
- To find the registered workflow, in the left-hand navigation bar, select “Public Favorites”. That listing will include the “WGS Processing Workflow” project. Open that project, and it will include the workflow “WGS processing workflow scattered over samples”. Open that workflow.
- Once you have found the registered workflow, you can run it your project by using the “Run Workflow” button in the toolbar and selecting your project (“WGS Processing Tutorial”) that you set up in Section 3a. Click OK to continue.
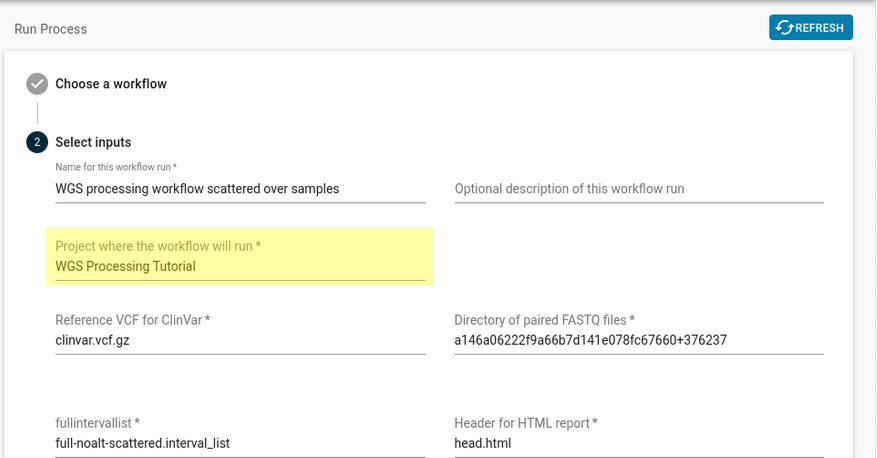
Figure 7: This is the dialog that pops up when you press “Run Workflow”. The project that you should select is highlighted in yellow. - Default inputs to the registered workflow will be automatically filled in. These inputs will work as-is, but can be edited. You can verify that they match the portable data hashes of the collections you copied over to your new project.
- Now, you can submit your workflow by selecting the “Run Workflow” button.

Congratulations! You have now submitted your workflow to run. You can move to Section 5 to learn how to check the state of your submitted workflow and Section 6 to learn how to examine the results of and logs from your workflow.
Let’s now say instead of running a registered workflow you want to run a workflow using the command line. This is a completely optional step in the walkthrough. To do this, you can specify cwl files to define the workflow you want to run and the yml files to specify the inputs to our workflow. In this walkthrough we will give two options (4b) and (4c) for running the workflow on the commandline. Option 4b uses a virtual machine provided by Arvados made accessible via a browser that requires no additional setup. Option 4c allows you to submit from your personal machine but you must install necessary packages and edit configurations to allow you to submit to the Arvados cluster. Please choose whichever works best for you.
4b. Optional: Setting up to Run a Workflow Using Command Line and an Arvados Virtual Machine
Arvados provides a virtual machine which has all the necessary client-side libraries installed to submit to your Arvados cluster using the command line. Webshell gives you access to an Arvados Virtual Machine (VM) from your browser with no additional setup. You can access webshell through the Arvados Workbench. It is the easiest way to try out submitting a workflow to Arvados via the command line.
New users are playground are automatically given access to a shell account.
Note: the shell accounts are created on an interval and it may take up to two minutes from your initial log in before the shell account is created.
You can follow the instructions here to access the machine using the browser (also known as using webshell):
Arvados also allows you to ssh into the shell machine and other hosted VMs instead of using the webshell capabilities. However this tutorial does not cover that option in-depth. If you like to explore it on your own, you can allow the instructions in the documentation here:
- Accessing an Arvados VM with SSH – Unix Environments
- Accessing an Arvados VM with SSH – Windows Environments
Once you can use webshell, you can proceed to section “4d. Running a Workflow Using the Command Line” .
4c. Optional: Setting up to Run a Workflow Using Command Line and Your Computer
Instead of using a virtual machine provided by Arvados, you can install the necessary libraries and configure your computer to be able to submit to your Arvados cluster directly. This is more of an advanced option and is for users who are comfortable installing software and libraries and configuring them on their machines.
To be able to submit workflows to the Arvados cluster, you will need to install the Python SDK on your machine. Additional features can be made available by installing additional libraries, but this is the bare minimum you need to install to do this walkthrough tutorial. You can follow the instructions in the Arvados documentment to install the Python SDK and set the appropriate configurations to access the Arvados Playground.
Once you have your machine set up to submit to the Arvados Playground Cluster, you can proceed to section “4d. Running a Workflow Using the Command Line” .
4d. Optional: Running a Workflow Using the Command Line
Now that we have access to a machine that can submit to the Arvados Playground, let’s download the relevant files containing the workflow description and inputs.
First, we will
- Clone the tutorial repository from GitHub (https://github.com/arvados/arvados-tutorial)
- Change directories into the WGS tutorial folder
$ git clone https://github.com/arvados/arvados-tutorial.git
$ cd arvados-tutorial/WGS-processing
Recall that CWL is a way to describe command line tools and connect them together to create workflows. YML files can be used to specify input values into these individual command line tools or overarching workflows.
The tutorial directories are as follows:
cwl– contains CWL descriptions of workflows and command line tools for the tutorialyml– contains YML files for inputs for the main workflow or to test subworkflows command line toolssrc– contains any source code necessary for the tutorialdocker– contains dockerfiles necessary to re-create any needed docker images used in the tutorial
Before we run the WGS processing workflow, we want to adjust the inputs to match those in your new project. The workflow that we want to submit is described by the file /cwl/ and the inputs are given by the file /yml/. Note: while all the cwl files are needed to describe the full workflow only the single yml with the workflow inputs is needed to run the workflow. The additional yml files (in the helper folder) are provided for testing purposes or if one might want to test or run an underlying subworkflow or cwl for a command line tool by itself.
Several of the inputs in the yml file point to original portable data hashes of collections that you make copies of in our New Project. These still work because even though we made copies of the collections into our new project we haven’t changed the underlying contents. However, by changing this file is in general how you would alter the inputs in the accompanying yml file for a given workflow.
The command to submit to the Arvados Playground Cluster is arvados-cwl-runner.
To submit the WGS processing workflow , you need to run the following command replacing YOUR_PROJECT_UUID with the UUID of the new project you created for this tutorial.
$ arvados-cwl-runner --no-wait --project-uuid YOUR_PROJECT_UUID ./cwl/wgs-processing-wf.cwl ./yml/wgs-processing-wf.yml
The --no-wait option will submit the workflow to Arvados, print out the UUID of the job that was submitted to standard output, and exit instead of waiting until the job is finished to return the command prompt.
The --project-uuid option specifies the project you want the workflow to run in, that means the outputs and log collections as well as the workflow process will be saved in that project
If the workflow submitted successfully, you should see the following at the end of the output to the screen
INFO Final process status is success
Now, you are ready to check the state of your submitted workflow.
5. Checking the State Of a Submitted Workflow
Once you have submitted your workflow, you can examine its state interactively using the Arvados Workbench. If you aren’t already viewing your workflow process on the workbench, you can navigate there via your project. You will want to go back to your new project, using the projects pulldown menu (the list of projects on the left) or searching for the project name. Note: You can mark a project as a favorite (if/when you have multiple projects) to make it easier to find on the pulldown menu by right-clicking on the project name on the project pulldown menu and selecting “Add to favorites”.
The process you will be looking for will be titled “WGS processing workflow scattered over samples” (if you submitted via the command line/Workbench).
Once you have found your workflow, you can clearly see the state of the overall workflow and underlying steps below by their label.
Common states you will see are as follows:
- “Queued” – Workflow or step is waiting to run
- “Running” or “Active” – Workflow is currently running
- “Complete” – Workflow or step has successfully completed
- “Failing”- Workflow is running but has steps that have failed
- “Failed”- Workflow or step did not complete successfully
- “Cancelled” – Workflow or step was either manually cancelled or was cancelled by Arvados due to a system error
Since Arvados Crunch reuses steps and workflows if possible, this workflow should run relatively quickly since this workflow has been run before and you have access to those previously run steps. You may notice an initial period where the top level job shows the option of cancelling while the other steps are filled in with already finished steps.
6. Examining a Finished Workflow
Once your workflow has finished, you can see how long it took the workflow to run, see scaling information, and examine the logs and outputs. Outputs will be only available for steps that have been successfully completed. Outputs will be saved for every step in the workflow and be saved for the workflow itself. Outputs are saved in collections. You can access each collection by clicking on the link corresponding to the output.

If we click on the outputs of the workflow, we will see the output collection. It contains the GVCF, tabix index file, and HTML ClinVar report for each analyzed sample (e.g., set of FASTQs). You can open a report in the browser by selecting it from the listing. You can also download a file to your local machine by right-clicking a file and selecting “Download” from the context menu, or from the action menu available from the far right of each listing.
Logs for the main process can be found back on the workflow process page by selecting the “LOGS” tab. You can view the logs directly through that panel, or in the upper right-hand corner select the “Outputs” button.
There are several logs available, so here is a basic summary of what some of the more commonly used logs contain. Let’s first define a few terms that will help us understand what the logs are tracking.
As you may recall, Arvados Crunch manages the running of workflows. A container request is an order sent to Arvados Crunch to perform some computational work. Crunch fulfils a request by either choosing a worker node to execute a container, or finding an identical/equivalent container that has already run. You can use container request or container to distinguish between a work order that is submitted to be run and a work order that is actually running or has been run. So our container request in this case is just the submitted workflow we sent to the Arvados cluster.
A node is a compute resource where Arvardos can schedule work. In our case since the Arvados Playground is running on a cloud, our nodes are virtual machines. arvados-cwl-runner (acr) executes CWL workflows by submitting the individual parts to Arvados as containers and crunch-run is an internal component that runs on nodes and executes containers.
stderr.txt- Captures everything written to standard error by the programs run by the executing container
node-info.txtandnode.json- Contains information about the nodes that executed this container. For the Arvados Playground, this gives information about the virtual machine instance that ran the container.
node.json gives a high level overview about the instance such as name, price, and RAM while node-info.txt gives more detailed information about the virtual machine (e.g., CPU of each processor)
- Contains information about the nodes that executed this container. For the Arvados Playground, this gives information about the virtual machine instance that ran the container.
crunch-run.txtandcrunchstat.txtcrunch-run.txthas info about how the container’s execution environment was set up (e.g., time spent loading the docker image) and timing/results of copying output data to Keep (if applicable)crunchstat.txthas info about resource consumption (RAM, cpu, disk, network) by the container while it was running.
usage_report.htmlcan be viewed directly in the browser by clicking on it. It provides a summary and chart of the resource consumption derived from the raw data incrunchstat.txt. (Available starting witharvados-cwl-runner2.7.2).container.json- Describes the container (unit of work to be done), contains CWL code, runtime constraints (RAM, vcpus) amongst other details
arv-mount.txt- Contains information using Arvados Keep on the node executing the container
hoststat.txt- Contains about resource consumption (RAM, cpu, disk, network) on the node while it was running
This is different from the log crunchstat.txt because it includes resource consumption of Arvados components that run on the node outside the container such as crunch-run and other processes related to the Keep file system.
- Contains about resource consumption (RAM, cpu, disk, network) on the node while it was running
For the highest level logs, the logs are tracking the container that ran the arvados-cwl-runner process which you can think of as the “workflow runner”. It tracks which parts of the CWL workflow need to be run when, which have been run already, what order they need to be run, which can be run simultaneously, and so forth and then creates the necessary container requests. Each step has its own logs related to containers running a CWL step of the workflow including a log of standard error that contains the standard error of the code run in that CWL step. Those logs can be found by expanding the steps and clicking on the link to the log collection.
Let’s take a peek at a few of these logs to get you more familiar with them. First, we can look at the stderr.txt of the highest level process. Again recall this should be of the “workflow runner” arvados-cwl-runner process. You can click on the log to download it to your local machine, and when you look at the contents – you should see something like the following…
2020-06-22T20:30:04.737703197Z INFO /usr/bin/arvados-cwl-runner 2.0.3, arvados-python-client 2.0.3, cwltool 1.0.20190831161204
2020-06-22T20:30:04.743250012Z INFO Resolved '/var/lib/cwl/workflow.json#main' to 'file:///var/lib/cwl/workflow.json#main'
2020-06-22T20:30:20.749884298Z INFO Using empty collection d41d8cd98f00b204e9800998ecf8427e+0
[removing some log contents here for brevity]
2020-06-22T20:30:35.629783939Z INFO Running inside container su92l-dz642-uaqhoebfh91zsfd
2020-06-22T20:30:35.741778080Z INFO [workflow WGS processing workflow] start
2020-06-22T20:30:35.741778080Z INFO [workflow WGS processing workflow] starting step getfastq
2020-06-22T20:30:35.741778080Z INFO [step getfastq] start
2020-06-22T20:30:36.085839313Z INFO [step getfastq] completed success
2020-06-22T20:30:36.212789670Z INFO [workflow WGS processing workflow] starting step bwamem-gatk-report
2020-06-22T20:30:36.213545871Z INFO [step bwamem-gatk-report] start
2020-06-22T20:30:36.234224197Z INFO [workflow bwamem-gatk-report] start
2020-06-22T20:30:36.234892498Z INFO [workflow bwamem-gatk-report] starting step fastqc
2020-06-22T20:30:36.235154798Z INFO [step fastqc] start
2020-06-22T20:30:36.237328201Z INFO Using empty collection d41d8cd98f00b204e9800998ecf8427e+0
You can see the output of all the work that arvados-cwl-runner does by managing the execution of the CWL workflow and all the underlying steps and subworkflows.
Now, let’s explore the logs for a subprocess in the workflow. Start by navigating back to the workflow process page. The logs can be found by selecting the appropriate subprocess under the “Subprocesses” tab, and getting the logs in the way as mentioned above. Let’s look at the log for the subprocess that does the alignment. That subprocess is named bwamem-samtools-view. We can see there are 10 of them because we are aligning 10 genomes. Let’s look at bwamem-samtools-view_2.
We click on the subprocess to open it and then can go down to the “Logs” section to access the logs. You may notice there are two sets of seemingly identical logs. One listed under a directory named for a container and one up in the main directory. This is done in case your subprocess had to be automatically re-run due to any issues and gives the logs of each re-run. The logs in the main directory are the logs for the successful run. In most cases this does not happen, you will just see one directory and one those logs will match the logs in the main directory. Let’s open the logs labeled node-info.txt and stderr.txt.
node-info.txt gives us information about detailed information about the virtual machine this step was run on. The tail end of the log should look like the following:
Memory Information
MemTotal: 64465820 kB
MemFree: 61617620 kB
MemAvailable: 62590172 kB
Buffers: 15872 kB
Cached: 1493300 kB
SwapCached: 0 kB
Active: 1070868 kB
Inactive: 1314248 kB
Active(anon): 873716 kB
Inactive(anon): 8444 kB
Active(file): 197152 kB
Inactive(file): 1305804 kB
Unevictable: 0 kB
Mlocked: 0 kB
SwapTotal: 0 kB
SwapFree: 0 kB
Dirty: 952 kB
Writeback: 0 kB
AnonPages: 874968 kB
Mapped: 115352 kB
Shmem: 8604 kB
Slab: 251844 kB
SReclaimable: 106580 kB
SUnreclaim: 145264 kB
KernelStack: 5584 kB
PageTables: 3832 kB
NFS_Unstable: 0 kB
Bounce: 0 kB
WritebackTmp: 0 kB
CommitLimit: 32232908 kB
Committed_AS: 2076668 kB
VmallocTotal: 34359738367 kB
VmallocUsed: 0 kB
VmallocChunk: 0 kB
Percpu: 5120 kB
AnonHugePages: 743424 kB
ShmemHugePages: 0 kB
ShmemPmdMapped: 0 kB
HugePages_Total: 0
HugePages_Free: 0
HugePages_Rsvd: 0
HugePages_Surp: 0
Hugepagesize: 2048 kB
Hugetlb: 0 kB
DirectMap4k: 155620 kB
DirectMap2M: 6703104 kB
DirectMap1G: 58720256 kB
Disk Space
Filesystem 1M-blocks Used Available Use% Mounted on
/dev/nvme1n1p1 7874 1678 5778 23% /
/dev/mapper/tmp 381746 1496 380251 1% /tmp
Disk INodes
Filesystem Inodes IUsed IFree IUse% Mounted on
/dev/nvme1n1p1 516096 42253 473843 9% /
/dev/mapper/tmp 195549184 44418 195504766 1% /tmp
We can see all the details of the virtual machine used for this step, including that it has 16 cores and 64 GIB of RAM.
stderr.txt gives us everything written to standard error by the programs run in this step. This step ran successfully so we don’t need to use this to debug our step currently. We are just taking a look for practice.
The tail end of our log should be similar to the following:
2020-08-04T04:37:19.674225566Z [main] CMD: /bwa-0.7.17/bwa mem -M -t 16 -R @RG\tID:sample\tSM:sample\tLB:sample\tPL:ILLUMINA\tPU:sample1 -c 250 /keep/18657d75efb4afd31a14bb204d073239+13611/GRCh38_no_alt_plus_hs38d1_analysis_set.fna /keep/a146a06222f9a66b7d141e078fc67660+376237/ERR2122554_1.fastq.gz /keep/a146a06222f9a66b7d141e078fc67660+376237/ERR2122554_2.fastq.gz
2020-08-04T04:37:19.674225566Z [main] Real time: 35859.344 sec; CPU: 553120.701 sec
This is the command we ran to invoke bwa-mem, and the scaling information for running bwa-mem multi-threaded across 16 cores (15.4x).
You can also view outputs for the subprocess just like you do for the main workflow process. Back on the subprocess page for bwamem-samtools-view_2, the Outputs pane shows the output files of this specific subprocess. In this case, it is a single BAM file. This way, if your workflow succeeds but produces a surprising result, you can download and review the intermediate outputs to investigate further.
We hope that now that you have a bit more familiarity with the logs you can continue to use them to debug and optimize your own workflows as you move forward with using Arvados if your own work in the future.
7. Conclusion
Thank you for working through this walkthrough tutorial. Hopefully this tutorial has helped you get a feel for working with Arvados. This tutorial just covered the basic capabilities of Arvados. There are many more capabilities to explore. Please see the links featured at the end of Section 1 for ways to learn more about Arvados or get help while you are working with Arvados.
If you would like help setting up your own production instance of Arvados, please contact us at info@curii.com.
Previous: Accessing Arvados Workbench Next: Getting started at the command line